
Les paramètres physico-chimiques sont très nombreux aussi bien beaucoup de techniques sont à disposition des laboratoires pour les étudier.
On veut
doser un élément ou plusieurs (ici le calcium Ca2+
et le magnésium Mg2+) dans leur matrice, leau.
On ajoute un indicateur
coloré qui va se lier avec lélément. Pour cette expérience,
on choisit le noir ériochrome (noté ID-) qui
passe de rouge à bleu en présence de Ca2+ et Mg2+.
On dose à laide de lEDTA ou acide éthylène diamine
tétra-acétique
.
Ca2+
et Mg2+ ont plus daffinité pour lEDTA que pour lID- ;
aussi, ils préfèrent laisser lindicateur coloré et se
lier avec lEDTA.
Quand on a autant
dEDTA que de Ca2+ et Mg2+, lID- est tout
seul dans la solution. Rappelons quil est bleu en présence du calcium
et du magnésium, coloration quil perd quand il est relargué,
pour redevenir rouge.
Concrètement,
on a équilibre des concentrations en Ca2+ / Mg2+
et dEDTA quand la solution est rouge.
|
La solution dEDTA est de concentration connue, ainsi on peut remonter à la concentration de Ca2+ / Mg2+ grâce à léquation CA.VA = CB.VB C
Concentration Application On
a [EDTA] = 100 mg/L. On dose 50 mL deau. La chute de burette est de 19.5
mL. ainsi CB = 39 mg/L Remarque : il faut ajouter une quantité très faible de noir ériochrome car il provoquerait une coloration trop intense du fait même de la couleur de la poudre. Ce phénomène rend difficile lobservation du changement de couleur. |
 |
Nous avons mentionné lajout dun indicateur coloré. Mais quest ce quun indicateur coloré ?
Il sagit dun acide faible ou dune base faible (donc faiblement dissociable dans le milieu aqueux) dont la forme acide et la forme basique ne sont pas de la même couleur.
![]()
En ajoutant
un indicateur en très faible quantité dans un milieu acide, celui-ci
est rouge alors quen milieu basique, il serait vert.
On veillera à
ne pas mettre trop dindicateur pour ne pas perturber les équilibres
de réaction. On met quelques gouttes diluées au centième
voire au millième, ou dans le cas dun indicateur solide, quelques microgrammes.
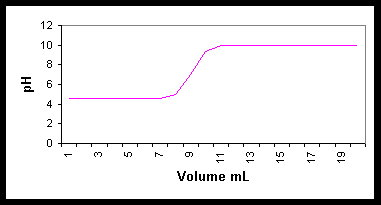
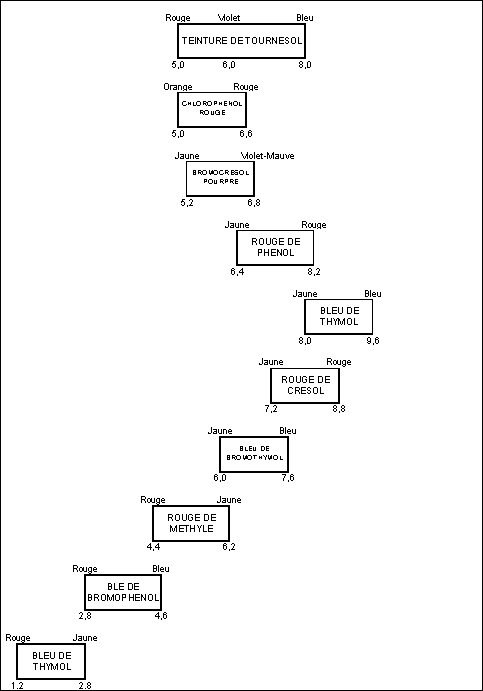
Pour choisir convenablement
son indicateur coloré, il faut connaître la courbe de neutralisation
de lélément que lon veut doser.
Alors, on choisira
un indicateur dont le virage coloré se situe sur le saut de pH de lélément
à doser.
Par exemple :
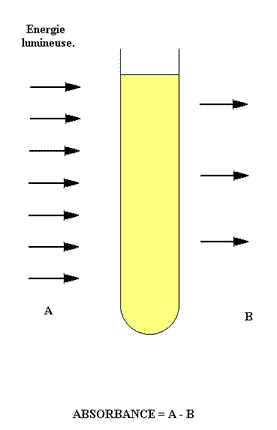 |
Chaque composé absorbe lénergie lumineuse à une longueur donde préférentielle que lon appelle pic dabsorbance. Le composé va retenir tout ou partie dun rayon lumineux.
|
 |
On peut également parlé de transmitance. De la même façon, on mesure larrivée de lénergie lumineuse par rapport à lémission et on a la quantité qui a été transmise => ce qui na pas été retenu par la solution.
Reprenons
le cas des agents de surface, on forme un complexe entre la molécule
dagent de surface et la molécule de bleu de méthylène.
On extrait le complexe de sa matrice grâce à du chloroforme (plus
grande affinité pour lui que pour leau en raison de la partie hydrophobe
de la molécule dagent de surface). Le bleu de méthylène
non complexé reste dans leau. Plus la couleur bleue du chloroforme est
prononcée, plus on a de complexe ce qui signifie que la concentration
en agents de surface était dautant plus importante dans leau.
Plus le complexe est
important dans la solution, plus labsorbance est grande : une grande partie
de lénergie lumineuse ne traverse pas la solution mais est absorbée
par le complexe.
A linverse, la transmitance
est de plus en plus faible avec laugmentation de la concentration en complexe.
La spectrométrie
est utilisée pour les nitrites avec lamino-4-benzène sulfonamide
(coloration rose) ou encore les phénols avec lamino-4-antipyrine (coloration
jaune là aussi avec une extraction chloroformique).
Il sagit
de techniques basées sur la séparation des composés. On
fait passer la matrice (eau) chargée de ces composés à
analyser à travers un support qui a la capacité de retenir plus
ou moins les solutés quelle contient.
Le support danalyse
est appelé phase stationnaire : elle ne bouge pas et sert
à entraver la migration des molécules et opérer une sélection
selon la taille et laffinité que les composés ont pour elle.
Selon sa nature, sa
structure chimique, ses groupes fonctionnels (alcool, acide
), chaque composé
réagira différemment.
Les composés
sont entraînés au moyen dun éluant : la phase mobile.
Il sagit dun liquide ou dun gaz qui circule dans la phase stationnaire et
qui va entraîner léchantillon. Les composés vont alors
traverser la phase stationnaire jusquà un détecteur. Celui-ci
réagit au passage de chaque composé.
Les solutés sont soumis à deux effets antagonistes :
Ils migrent
alors selon un ordre les uns par rapport aux autres et un temps dictés
par le système chromatographique (phase stationnaire + phase mobile).
Les deux phases ne
se mélangent jamais. La phase stationnaire est insoluble pour
garder lintégrité du système.
On utilise différentes
techniques en chromatographie. En voici quelques unes.
La Chromatographie
Liquide Haute Performance est la technique la plus utilisée dans
les laboratoires car elle est la plus facilement adaptable en routine, facile
à manipuler et un très grand nombre de composés se prêtent
à ce système.
Elle est principalement
utilisée pour les molécules de volume important comme les
pesticides ou les molécules biologiques (que nous naborderons pas dans
cet exposé). Ce sont des molécules complexes qui ont un encombrement
stérique (= volume, organisation spatiale de la molécule). On
lutilise pour les composés thermolabiles = qui ne supportent pas la
chaleur (on ne peut pas les analyser par Chromatographie en Phase Gazeuse).
Les mécanismes de rétention sont régis par plusieurs phénomènes :
- Lencombrement stérique. Plus la molécule est grosse et " tordue ", plus elle aura du mal à migrer dans la colonne au travers des pores de la phase stationnaire.
- La différence de solubilité des composés dans les phases stationnaire et mobile. Si on a une phase stationnaire non polaire (ex : greffon organique) et une phase mobile polaire (eau). On veut rechercher une molécule organique : elle aura plus daffinité pour le greffon organique que pour leau. La molécule est attirée par la phase stationnaire et repoussé par la phase mobile. La rétention est donc importante.
Dans le
même sens, on peut avoir un effet de répulsion du fait, par exemple,
du caractère hydrophobe des molécules. Celles-ci restent à
proximité de la phase stationnaire pour " fuir "
la phase mobile à majorité deau. Néanmoins, elles subissent
tout de même sa force délution et migrent mais très lentement.
On peut avoir une
augmentation du temps délution quand les molécules arrivent à
" saccrocher " par des liaisons faibles dites liaisons
hydrogène. Elles sont " liées " à la
phase stationnaire et la phase mobile mettra beaucoup de temps à les
entraîner tout au long de la colonne.
Lappareillage.
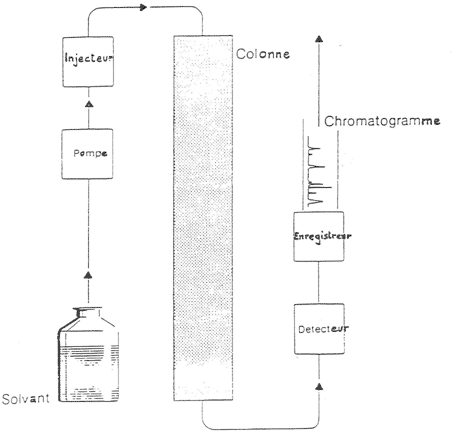
Le solvant. Dans le jargon de chimiste, on parle de phase mobile ou déluant. Une pompe assure un débit constant de la phase mobile à travers tout le système.
Linjecteur permet dintroduire un volume TRES précis de solution à analyser. Ce volume est de lordre de 40 µL soit 0.04 mL. Ce volume nest pas fixe mais peut varier selon lappareillage utilisé.
La colonne, ou phase stationnaire, qui sert à la séparation des composés est assez courte : de lordre de 20 cm la plupart du temps.
Le détecteur
permet de repérer le passage dun composé et den informer lenregistreur.
Celui-ci va stocker les données du détecteur et les restituer
sous la forme dun graphique : un chromatogramme. On a différent
type de détecteur. Le plus couramment utilisé est le détecteur
à U.V.. Il fonctionne selon les absorbances. Son inconvénient
est que les composés doivent absorbés dans lU.V. Par exemple,
les alcanes (chaîne carbonée simple) ne sont pas repérés.
Il présente
toute fois lavantage dêtre très stable au niveau signal de détection.
Le détecteur
universel est le détecteur différentiel. Il mesure la différence
de réfraction entre la phase mobile pure et léluat. Il est très
sensible aux variations de température et de débit de phase mobile.
Les phases stationnaires.
En chromatographie, on utilise de la silice sous forme de gel de silice poreux. La phase stationnaire est globalement insoluble. On dit globalement car il arrive toujours que des molécules se " détachent " de leur support pour se solubiliser. Ce phénomène est toutefois négligeable et souvent ces molécules retournent sur la silice avant de sortir de la colonne.

Selon cette
structure, le site OH est le site actif : il provoque la rétention
en attirant les molécules.
Lhumidité
contenue dans la silice est très difficilement maîtrisable et mesurable.
Cela est gênant car leau est une molécule polaire, elle peut influencer
les molécules passant à proximité et, dans notre cas, perturber
leur temps de rétention.
Pour éviter
cela, on utilise de plus en plus de la silice greffée. Sur loxygène
de Si-OH, on greffe une molécule qui nest pas polaire et rendra plus
facile la maîtrise de la manipulation. On utilise des molécules
de taille variable. Elles peuvent être composées de 2 à
18 atomes de carbone voire même plus.
La greffe se fait de la manière suivante.
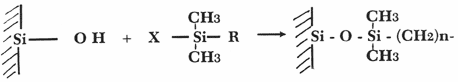
On dit que
lon a des phases inversées en raison du déplacement de
la polarité dans le système.
En " phases
normales ", la silice est humide ce qui la polarise. On utilise alors
un éluant non polaire comme le méthanol. Dans le cas de phases
inversées, la phase stationnaire porte un greffon qui ne peut accepter
de molécule deau, elle est donc non polaire. On utilise alors une phase
mobile polaire : leau.
En réalité,
on ne choisit pas entre les deux mais on réalise un mélange selon
les besoins de lanalyse (cf. Optimisation des conditions danalyse).
Une des autres caractéristiques
de la colonne est sa granulométrie, cest à dire la taille
de ces pores. On peut imager en disant que la phase stationnaire est une ensemble
de petites billes de diamètre variable selon la colonne, de 5 à
10 µm. Plus cette granulométrie est faible, plus les pics du chromatogramme
sont fins. En effet, les pores qui pourraient retenir les molécules sont
petits, ainsi les molécules ne peuvent pas y rentrer et sy " coincer ".
Elles migrent donc à peu près toutes en même temps.
Les phases mobiles.
Rappelons
que la phase mobile sert à entraîner, à débit constant,
les composés à travers la colonne.
En HPLC, on utilise
souvent un mélange méthanol/eau dans des proportions variables
que lon adapte selon les besoins de lanalyse.
Il est capital que
le débit soit constant afin de ne pas faire subir aux composés
une force délution variable. Cela pourrait jouer sur le temps délution.
Dans le cas de chromatographie
à polarité de phases normale, le composant majoritaire dans léluant
sera le solvant organique (dans la plupart des cas, le méthanol) et de
leau dans le cas de chromatographie à polarité de phases inversée.
Sur les documents
danalyse, on note la composition de la phase mobile comme suit :
MeOH/H2O 70/30
Cela signifie que le mélange est composé de 70% de méthanol et de 30% deau.
Optimisation des conditions danalyse
Lors des
analyses chromatographiques, les deux problèmes majeurs rencontrés
sont :
- Le temps
délution qui peut être trop long dun point de vue
adaptation en routine.
- La mauvaise
séparation de deux ou plusieurs composés.
Pour remédier à ces deux problèmes principaux, on a recourt à diverses techniques :
Lorsque
le temps délution doit être réduit, on augmente le débit
de la phase mobile afin daccroître sa force délution et ainsi
daccélérer la sortie des composés.
Cette technique est
très utilisée mais a ces limites. On ne peut pas augmenter infiniment
de débit. Il faut tenir compte des capacités de lappareillage.
De plus, il y a une limite au delà de laquelle il est physiquement impossible
daller en fonction de lappareillage. Cest un problème de pression
engendrée alors dans le système. Lors de la mise au point de la
technique danalyse, on réalise une série dessais visant à
déterminer le débit optimal.
Lorsque lon recherche plusieurs composés, il peut se produire une sortie quasi simultanée de tous ces composés lorsque le débit est trop important. On a alors un chevauchement des pics voire confusion. Lintégrateur (partie de lenregistreur) est alors incapable de calculer de façon correcte laire du pic et donc, on ne peut en déduire la concentration des composés dans la solution
.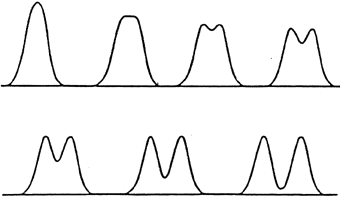
En effet,
pour pouvoir déterminer les concentrations dans les solutions, il faut
quil y ai retour à la ligne de base (pas de détection de composé ;
Etat " zéro ") entre deux pics. Ceux-ci doivent être
indépendants et délimités précisément. Sans
quoi, lintégrateur ne peut donner une valeur précise et exploitable
de laire du pic.
Quand on veut réduire
le temps délution, on augmente le débit de la phase mobile. Et
bien, à linverse, quand on a un chevauchement de pic, on diminue ce
débit pour réduire la force délution. Ainsi les composés
" prennent leur temps " pour sortir. Il y a bonne séparation
de ceux-ci dans la colonne.
Il faut savoir ajuster
le débit afin dobtenir le meilleur résultat possible.
Par exemple,
on va baisser la fraction deau de léluant pour faciliter la migration
dun composé hydrophobe. On le rejette moins vers la phase stationnaire
donc sa migration est facilitée.
Lors de lutilisation
de la nouvelle phase mobile, il faut prendre garde à bien rincer la colonne
pour se mettre dans les bonnes conditions expérimentales et ne pas avoir
un mélange des deux phases mobiles sans quoi lexploitation des résultats
en devient difficile.
Il sagit
dune solution de dernier recourt car cest une opération longue. Lancienne
colonne doit être rincée avec un grand volume déluant afin
de bien enlever toute trace de solution antérieurement analysée.
Le détecteur permet de sassurer quelle est bien propre. Lors de linstallation
de la nouvelle colonne, il faut là encore faire passer un volume important
de phase mobile afin de la remplir. Dans le cas où cette manipulation
est mal faite, on peut avoir des problèmes car léchantillon remplirait
ces vides. Là encore les résultats seraient faussés car
il faudrait plus de temps pour éluer tous les composants.
On préconise
de faire circuler dix fois le volume de la colonne pour assurer le meilleur
lavage et remplissage possible.
Exploitation des résultats.
Les résultats sont fournis pas lenregistreur sous la forme dun chromatogramme.

A chaque pic de la courbe correspond la sortie dun composé.
Toutes les molécules dun soluté ne sortent pas simultanément. Il existe de nombreux trajets de longueurs différentes au travers de la colonne.

Plus le
débit de la phase mobile sera faible, plus les pics auront une base large
du fait même de la faible force délution du solvant.
Ce que lon appelle
ligne de base correspond à la détection de la phase mobile seule.
Au début de la manipulation , on fait passer de la phase mobile seule
et on informe le détecteur quil sagit du " zéro "
de détection. Cest la ligne de base.
Le chiffre inscrit
en haut du pic indique le temps délution (à la pointe du pic)
pour chacun des composés.
Ce chiffre permet
de retrouver, dans le listing des résultats, laire du pic correspondante.
Pour doser un composé
de concentration inconnue dans une solution, on établit tout dabord
une gamme détalonnage.
Le principe
est le suivant :
On injecte des solutions
de concentrations connues que lon élue les unes après les autres
afin de connaître la correspondance concentration ó aire du pic.
Dans lidéal,
on essaie davoir une gamme détalonnage qui encadre la valeur présumée
de léchantillon à doser. Ainsi on sassure que cette valeur est
bien dans le domaine de linéarité de la droite détalonnage
et que lon pourra utiliser léquation de la droite pour trouver la concentration
inconnue.
Léquation
de la droite détalonnage est de la forme y = ax + b
Avec y aire
du pic
x concentration
dans la solution
a pente de la droite
b ordonnée
à lorigine (égale à zéro car si on na pas de composé,
on na pas de pic).

Connaissant
laire du pic de la solution inconnue et la pente de la droite étalon,
il est facile de remonter à la concentration de la solution inconnue.
Parfois, il arrive
que les résultats soient totalement inexploitables.
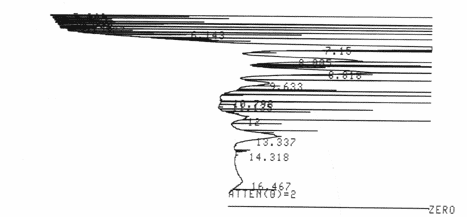
Ici, on
a saturation de la colonne. La concentration de la solution était
trop importante et on a rempli la colonne, elle est saturée. Il faut
alors la laver avec de la phase mobile pure jusquà ne plus obtenir de
signal sur le chromatogramme : la colonne est alors propre et on peut recommencer
avec un échantillon dilué.
On peut aussi avoir
le cas dune pollution de la colonne lorsque la solution est trop chargée
déléments divers. Ils interférent tous entre eux et aucune
aire de pic nest exploitable. Dans ce cas-là, on purifie un maximum
la solution grâce à des résines. Elles ont la capacité
de retenir un élément, ou famille déléments, spécifique.
On retire ainsi un maximum dinterférents dans le but de doser la solution
dans des conditions acceptables en éliminant les chevauchements de pics.
Il est à noter
que le détecteur a ces limites. Il ne peut pas déceler de composés
en dessous dune certaine concentration, variable selon la performance du matériel.
On parle de limite de détection.
Bien que
lHPLC soit très répandue, il existe dautres techniques chromatographiques
qui connaissent quelques variantes par rapport à lHPLC.
Voyons ce quil en
est de la CPG.
La Chromatographie en Phase Gazeuse repose sur le même principe que lHPLC. Cependant, la phase mobile est gazeuse et on travaille à haute température (de lordre de 80°C).
Lappareillage.
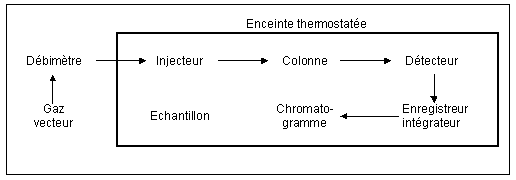
Le gaz
vecteur est la phase mobile puisque cest lui qui va éluer la solution
tout au long de la colonne.
Le débitmètre
contrôle lapport continu et régulier de la phase mobile. Il peut
également être présent en HPLC.
La colonne
conserve son rôle de rétention des composés en vue de leur
séparation.
Le détecteur
qui sert à repérer le passage dun composé. Ces données
sont transmises à lenregistreur-intégrateur qui produit le chromatogramme.
Le fonctionnement est le suivant.
On introduit une très faible quantité de mélange homogène à analyser dans linjecteur. Ce dernier permet à la fois de porter léchantillon à létat de vapeur mais aussi de lintroduire dans le flux de léluant. Linjection doit être rapide afin de ne pas vaporiser léchantillon dans la seringue qui sert à introduire léchantillon dans linjecteur. Dans le cas contraire, on aurait des pertes de matière et donc des résultats erronés.
Lensemble du système chromatographique, de linjecteur à lenregistreur, est à une température suffisante pour maintenir léchantillon à létat de vapeur. Cette température est variable dune analyse à lautre. Par exemple, pour les hydrocarbures, la colonne est à 100°C, linjecteur est le détecteur sont à 150°C. Lentrée et la sortie du système sont à des températures plus élevées que la colonne afin dêtre certain davoir un échantillon à létat de vapeur.
Alors que lHPLC na besoin que dun liquide vecteur, la CPG a besoin de trois gaz. Un qui élue léchantillon et deux qui font fonctionner le détecteur.

Prenons lexemple du FID. Le Détecteur à Ionisation de Flamme fonctionne avec de lair et de lhydrogène.
Ce détecteur est spécifique des molécules organiques.
Il existe
de nombreux autres détecteurs comme le détecteur à conductivité
thermique (qui a le gros avantage dêtre universel) ou encore à
capture délectrons (système radioactif nécessitant une
autorisation gouvernementale).
La CPG est très
performante mais elle a le gros inconvénient de nécessiter
une température de travail élevée. On ne peut donc
pas analyser par ce système les molécules dites thermolabiles :
celles qui ne supportent pas la chaleur. Elles seraient détruites par
la chaleur. Citons comme exemple les vitamines.
Les phases stationnaires.
En CPG,
les mécanismes de rétention sont moins efficaces quen HPLC aussi
on utilise des colonnes beaucoup plus longues : de 20 à 25 m.
On distingue trois
types de colonnes :
-- colonne remplie
ou à garnissage. Le " tube " est rempli dun
support (polymère
) sur lequel on fixe la phase stationnaire (silice,
silice greffée). Le diamètre intérieur est de 2 à
3 mm.
-- colonne capillaire.
Le tube est très fin. Le diamètre intérieur est inférieur
à 0.53 mm. Les parois internes sont tapissées de phase stationnaire
(silice ou silice greffée).
-- colonne macrobore,
dont le diamètre intérieur est supérieur à 0.53
mm mais inférieur à 2 mm. Selon le même principe que pour
les autres colonnes, lintérieur est tapissé avec la phase stationnaire.
On peut envisager :
-- une phase stationnaire liquide que lon fixe sur un support
Cas de la colonne remplie avec un support sous forme de grains.
Cas de la colonne capillaire et macrobore si on tapisse la surface interne du tube.
Remarque :En CPG comme en HPLC, quand on parle phase stationnaire liquide, elle est en fait très visqueuse pour ne pas se mêler à la phase mobile.
-- une
phase stationnaire solide : il peut sagir de granules dont on va remplir
la colonne ou encore un revêtement poreux sur la surface interne dune
colonne capillaire ou macrobore.
On a ici le même
système quen HPLC à savoir que la silice peut être
simple ou greffée avec des groupements en carbone de longueur
variable.
La granulométrie
est de plusieurs centaines de µm. On dit alors que la colonne est peu efficace
car elle produit des pics larges (selon le même principe que décrit
pour lHPLC).
Les phases mobiles.
Ici, on a un gaz vecteur comme éluant de léchantillon. Il peut sagir dhexane (molécule linéaire de formule brute C6H14), de diazote N2 ou de dihydrogène H2. Le débit est fortement variable en fonction de la colonne : environ 30 mL/min pour une colonne remplie et environ 1 mL/min pour une colonne capillaire. Cette importante différence sexplique principalement par la différence de diamètre intérieur des colonnes.
Optimisation des conditions danalyse.
En plus
des réglages de débit de la phase mobile que nous avons vus en
HPLC, on peut agir sur la température du four qui contient le
système chromatographique.
Lintérêt
de travailler à une température élevée est que lon
a des molécules excitées. Elles ont donc des liaisons hydrogènes
(liaisons qui font que les matières peuvent être à létat
liquide, solide ou gazeux selon la température et la pression) très
lâches. Elles ne sont pas très attirées les unes par rapport
aux autres. Les pics des chromatographes sont dautant plus étendus car
les molécules narrivent pas en " masse " mais de
façon assez dispersée.
Quand on baisse la
température, on augmente la force des liaisons hydrogène entre
les molécules et ainsi la " cohésion " du
groupe de molécules de même nature.
Rappelons que la largeur
des pics nest pas dictée par la seule température de travail
et le débit de léluant. Tout comme en HPLC, la granulométrie
joue un rôle capital. Plus les pores sont gros, plus ils peuvent loger
les molécules et les retenir.
Exploitation des résultats.
Tout comme
pour lHPLC, on a des résultats sous forme de chromatogramme.
On exploite les résultats
selon le même principe.
Cest aussi
une technique chromatographique. Cependant elle est basée sur le caractère
électronégatif des éléments.
Agée de
moins de dix ans, cette technique propose une rapidité de séparation,
une qualité danalyse ainsi quun emploi facile. Elle rivalise aisément
avec les techniques classiques didentification des ions.
Principe.
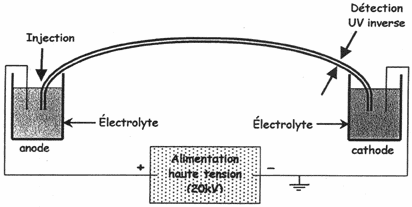
Une différence
de potentiel de 20kV est appliquée aux électrodes plongeant
dans les deux réservoirs remplis de la même solution aqueuse que
le capillaire. Lintensité ne doit pas dépasser 100 µA pour ne
pas provoquer un échauffement du capillaire.
Les ions migrent selon
deux phénomènes.
- Lélectromigration :
dans toute expérience délectrophorèse, un composé
porteur dune charge électrique migre, sous linfluence du champ électrique
appliqué E, à une vitesse qui est la vitesse de migration électrophorétique ;
cette vitesse dépend de la charge de lion, de ses dimensions et de la
viscosité de lélectrolyte.
- Mobilité
électro-osmotique, flux électro-osmotique : le capillaire
étant fait de silice, ses parois sont recouvertes de sites silanols (comme
en HPLC et CPG) Si-OH qui peuvent sioniser si le pH est supérieur à
2 ; le tube du capillaire est alors recouvert dune couche fixe polyanionique
(plusieurs anions) et polycationique (positive) qui se met en mouvement sous
linfluence de la différence de potentiel et migre alors vers la cathode.
Ce déplacement est appelé le Flux Electro-Osmotique ou FEO.
Ce flux permet dentraîner toutes les molécules et éléments
présents dans le capillaire sans en modifier lordre de sortie et la
séparation.
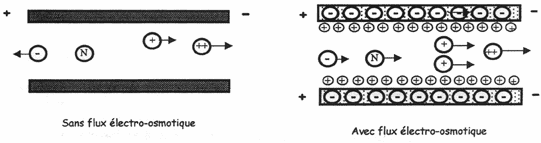
Dans cette technique, on utilise le FEO dans le but dimposer un sens de migration à lensemble des espèces chargées de léchantillon. Les espèces non chargées " suivent la marche ".
Le passage
de lélectrolyte (solution servant à entraîner les composés)
est possible grâce à un système daspiration. Une des deux
extrémités du capillaire est plongée dans le liquide, on
aspire à lautre bout du capillaire. Ce système est appelé
linjection hydrostatique. On peut appliqué le principe inverse
en effectuant une pression sur la solution à analyser pour la " pousser "
dans le capillaire.
Concernant le système
de détection, on applique la détection inverse.

Tout comme en HPLC, le détecteur mesure labsorbance, dans la zone U.V. de la solution. Toutefois, on observe une variante. Les anions ont une très faible absorbance, il faut donc choisir un électrolyte qui aura des propriétés absorbantes importantes (exemple : le chromate).
La présence des composés dans lélectrolyte va induire un phénomène de dilution de celui-ci. De cette façon, labsorbance de lélectrolyte dans lU.V. va fortement diminuée au passage des composés. Ainsi on a des pics " négatifs " sur lenregistrement car le " zéro " est fait sur lélectrolyte seul. Les appareils modernes permettent de demander une représentation " classique " des résultats.
Certains paramètres ont des techniques qui ne sappliquent quà eux. Parmi ces mesures, les plus couramment utilisées sont les MES, la DCO et la DBO. Ces données étant très fréquentes dans le domaine de lanalyse de leau et notamment pour le contrôle des eaux usées, nous exposerons plus spécifiquement leur principe général.
Les MES sont de deux natures. Organique et minérale.
Voici le principe dobtention des MES organiques.
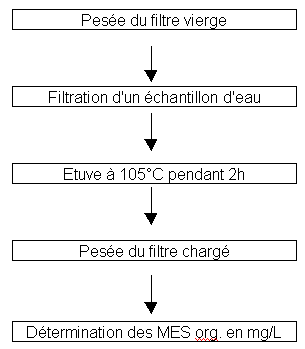
Les matières minérales sobtiennent selon le même schéma. Le passage au four est plus long et plus chaud dans le but de détruire la matière organique pour ne garder que les matières minérales.
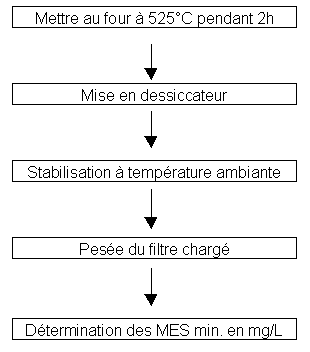
Le dessiccateur permet de stabiliser lhumidité dans léchantillon.
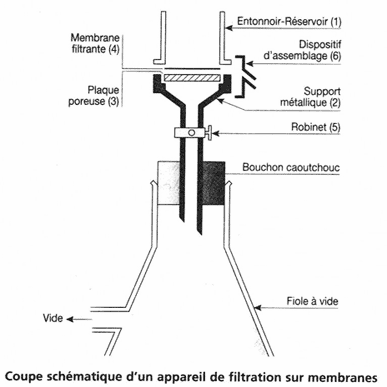
La filtration se fait de la manière suivante.
Cette donnée est obtenue en deux étapes bien distinctes.
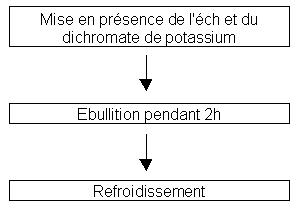
Le dichromate de potassium est un puissant oxydant qui va sattaquer à toute la matière organique de léchantillon
Selon le principe du dosage acido-basique, on va doser lexcès de dichromate de potassium : la quantité qui na pas réagi avec la matière organique de léchantillon ; cela grâce à la solution de sel de Mohr. On connaît la quantité initiale de dichromate de potassium, on peut donc remonter à la quantité de dichromate oxydée.
La DBO5

On mesure la quantité doxygène dégradé au travers de processus chimique et biologique. On utilise des sondes qui mesurent la pression en oxygène. Elles rendent directement compte de la quantité doxygène consommée en mg/L cela grâce à une différence de pression dans le flacon.
Lors de la dégradation, du CO2 est produit. Pour éliminer cette interférence, on place 2 à 3 pastilles de soude dans le flacon de mesure. Elle a pour rôle de piéger le CO2.